19 sept. 22

Tiempo estimado de lectura:min
Le méthylome, nouvelle carte d’identité (épi)génétique des cancers
Audrey Rousseau, Université d'AngersMieux comprendre et traiter le cancer nécessite de décrypter les rouages de cette maladie – ou plutôt de ces maladies, tant il peut prendre des formes multiples.
Un moyen en plein développement est de se pencher sur l’ADN. Toutes nos cellules (y compris les cellules tumorales) ont, dans leur noyau, une molécule d’ADN : sorte de long texte où s’enchaînent notamment les gènes et toute l’information génétique qui contribue à nous définir.
Les cancers résultent de modifications de ce texte génétique (on parle de mutations de sa « séquence ») survenant dans une cellule, qui peuvent être particulièrement massives et entraîner sa multiplication incontrôlée. Se développe alors une tumeur qui, alors qu’elle grandit et évolue, peut envahir l’organisme.
Les progrès récents des techniques de séquençage « à haut débit » (permettant la lecture rapide du texte génétique) ont permis d’identifier les modifications se cachant derrière de nombreux cancers. Désormais, il est possible de répertorier le « catalogue » de mutations d’une tumeur afin d’établir sa carte d’identité : un atout considérable pour mieux appréhender la nature du cancer en question, ses origines, ses rouages internes et son pronostic. Or une meilleure compréhension de la maladie contribue au développement de traitements plus efficaces.
Un second niveau d’information génétique
Mais l’information génétique ne se situe pas que dans le texte inscrit dans notre ADN… Un second niveau de codage, dit « épigénétique », a été identifié, dont l’étude s’est développée ces dernières décennies. L’épigénétique est à la génétique ce que la ponctuation est à une phrase : le sens d’une phrase sera différent selon la présence et la localisation de virgules, tirets ou parenthèses…
Les modifications épigénétiques (la ponctuation) de l’ADN (les phrases) sont ainsi capables d’influer le message codé dans l’ADN, et sur la façon dont il est exprimé.
D’un point de vue chimique, nos « virgules génétiques » peuvent prendre la forme d’ajout (ou de retrait) de groupements d’atomes spécifiques – en l’occurrence des méthyls (CH3). La lecture de ce « code » épigénétique permet d’établir un autre type de carte d’identité : le méthylome.
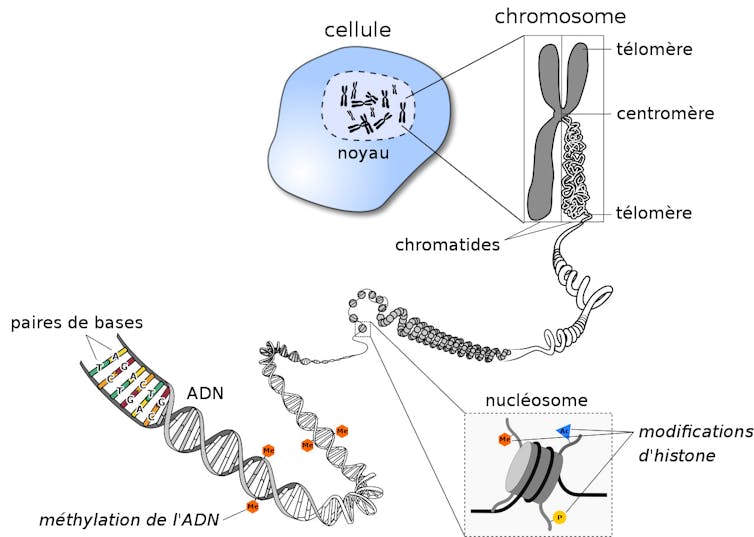
Là encore, son étude fine et sa comparaison avec des méthylomes de cellules saines et tumorales sont riches en informations : si la lecture de la séquence (du texte) génétique renseigne sur les mécanismes de croissance exubérante de la cellule cancéreuse, celle du méthylome précise la nature du cancer et son origine. In fine, le décryptage de ces deux niveaux de données complémentaires contribue à mieux caractériser le cancer et à choisir les traitements anticancéreux les plus adaptés.
Un usage pionnier en neuro-oncologie
Si les techniques de séquençage à haut débit ont pris leur envol à la fin du XXe siècle, l’étude du méthylome appartient clairement au XXIe siècle.
Du fait de son coût et du matériel nécessaire pour cartographier la ponctuation de l’ADN, cette technique n’est pour l’heure mise en place que dans certains centres d’oncologie médicale spécialisés. En clinique, c’est surtout en neuro-oncologie (tumeurs du cerveau et de la moelle), pionnière dans le domaine, que son utilisation est la plus aboutie – notamment dans les cas de diagnostic difficile. L’Organisation mondiale de la santé (OMS) recommande d’ailleurs son étude dans le diagnostic de nombreuses tumeurs cérébrales depuis 2021.
Si d’autres types de cancer (sarcomes, développés à partir des os, des muscles, de la graisse…) commencent également à être étudiés par leur méthylome, les résultats sont encore préliminaires.
L’analyse des données de méthylome repose sur la bio-informatique et nécessite le développement d’algorithmes (formules de calcul) d’intelligence artificielle (IA). L’idée de base est simple : il faut classer ensemble les tumeurs qui ont la même ponctuation (donc les mêmes variations au niveau épigénétique), à l’instar d’un jeu de 7 familles où le joueur cherche à rassembler les individus d’une même famille.
[Plus de 80 000 lecteurs font confiance à la newsletter de The Conversation pour mieux comprendre les grands enjeux du monde. Abonnez-vous aujourd’hui]
Une large base de données développée par une équipe de l’université d’Heidelberg rassemble déjà les profils de méthylation de presque 100 000 tumeurs cérébrales, classées en 80 familles ou sous-familles.
En libre accès, elle permet l’envoi de nouvelles données qui sont analysées en ligne gratuitement. L’avantage est double puisque d’une part le logiciel propose au médecin pathologiste un classement de la tumeur envoyée, et l’équipe d’Heidelberg enrichit sa base de données, rendant les algorithmes d’IA plus performants – de façon générale, plus un algorithme analyse de tumeurs, plus les classements qu’il propose seront fiables. Cette forme de projet collaboratif international bénéficie ainsi au plus grand nombre : patients, médecins et chercheurs.
Dans certains cas, la machine peut se révéler meilleure que le médecin pathologiste qui, de façon traditionnelle, examine les cellules cancéreuses au microscope. Mais si la machine peut parfois dépasser l’humain, elle peut aussi être prise en défaut face à des tumeurs très rares qu’elle n’aurait pas (ou peu) rencontrées : le risque est alors qu’elle ne réussisse pas à classer la tumeur ou, plus grave, la classe dans une mauvaise famille. C’est pourquoi tout diagnostic est vérifié par un médecin pathologiste, qui réalise une synthèse de son diagnostic microscopique, des résultats du méthylome et des mutations détectées à la lecture du texte génétique.
Tout diagnostic final est ainsi dit « intégré » car il prend en compte les données microscopiques, génétiques et épigénétiques. Plus fiable, il permet à l’oncologue de choisir au mieux les traitements.
Malgré les limites de l’IA, l’analyse du méthylome représente bien un progrès considérable dans le diagnostic des tumeurs cérébrales, notamment de l’enfant – chez qui elles sont beaucoup plus variées que chez l’adulte. En France, les Centres hospitalo-universitaires (CHU) référents en neuro-oncologie s’équipent progressivement afin que, dans les années à venir, une carte d’identité épigénétique puisse être établie pour chaque tumeur cérébrale (en plus de la carte d’identité génétique déjà réalisée).
Une technique qui va encore gagner en puissance
Contrairement au texte génétique qui est fixé littéralement dans l’œuf, dès notre conception, et est très complexe à modifier, l’information épigénétique est plus « modulable ». Certains traitements appelés « épidrogues » peuvent ainsi changer la ponctuation de l’ADN et contribuent à freiner l’évolution du cancer. Si la recherche à leur sujet est toujours en cours pour comprendre leur impact global sur le corps, ils pourraient être administrés en complément des traitements conventionnels (radiothérapie et chimiothérapie).
En oncologie, la notion de « médecine personnalisée » ou « médecine de précision » (traitements sur mesure adaptés à la personne et spécifiques du type de cancer) est sur toutes les lèvres. En effet, les chimiothérapies très générales ne sont pas efficaces chez tous les patients et touchent aussi les tissus sains, y provoquant de nombreux effets indésirables. Choisir des médicaments à partir des caractéristiques génétiques et épigénétiques du cancer donne l’espoir de voir apparaître un meilleur contrôle de la maladie et d’épargner, dans une certaine mesure, les tissus sains (avec des « thérapies ciblées »).
Ainsi, le méthylome semble avoir de beaux jours devant lui. Même si la ponctuation n’apparaît pas, au premier abord, essentielle à une phrase, elle est indispensable à la lecture d’un texte et à sa bonne compréhension. On sait aujourd’hui que le cancer est une maladie génétique et épigénétique et que les virgules comptent autant que les consonnes et les voyelles.![]()
Audrey Rousseau, Professeur en Anatomie Pathologique - Médecin enseignant-chercheur au CHU d'Angers, Université d'Angers
