14 Nov 22
Geschätzte Lesezeit:min
Une nouvelle théorie étonnante sur les causes de la maladie d’Alzheimer
En 1906, le psychiatre et neuroanatomiste Alois Alzheimer a rapporté « un processus pathologique grave et particulier dans le cortex cérébral » lors d’une réunion de psychiatres à Tübingen, en Allemagne. Le cas décrit était celui d’une femme de 50 ans qui souffrait de pertes de mémoire, de délires, d’hallucinations, d’agressivité et de confusion – des troubles qui se sont tous aggravés jusqu’à son décès prématuré, survenu cinq ans plus tard.
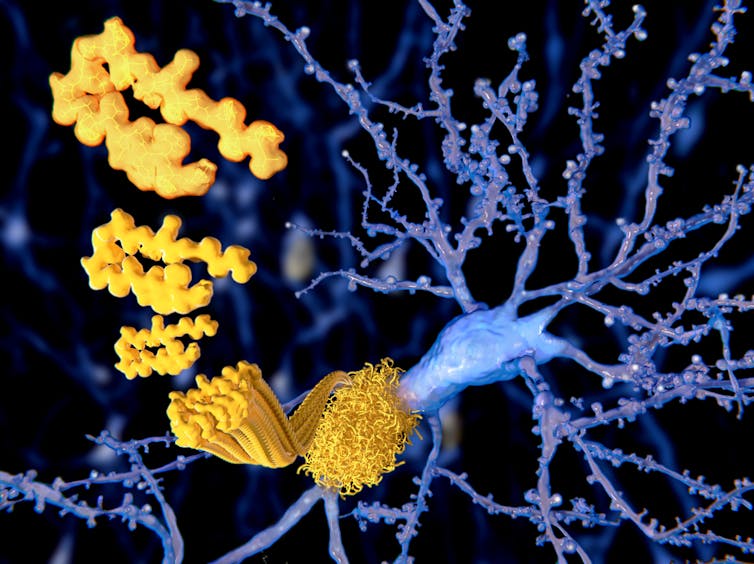
Lors de l’autopsie, Alzheimer a remarqué la présence de plaques dans le cerveau de la patiente. Ces plaques – des agrégats de protéine amyloïde bêta – sont encore aujourd’hui considérées comme étant responsables de la maladie d’Alzheimer.
Cette théorie comporte toutefois deux lacunes. Premièrement, elle n’explique pas pourquoi de nombreuses personnes (dont certaines sont âgées) présentent des plaques au cerveau sans pour autant souffrir de symptômes neurologiques, tels que la perte de mémoire. Deuxièmement, les essais cliniques de médicaments qui réduisent ces plaques ont été infructueux – sauf pour une exception récente, à laquelle nous reviendrons plus loin.
Lorsque la protéine amyloïde bêta s’accumule sous forme de plaques (agrégats insolubles), la forme soluble originale de la protéine, qui remplit des fonctions importantes dans le cerveau, est consumée et perdue. Certaines études ont révélé que la réduction de la quantité d’amyloïde bêta soluble – appelée amyloïde bêta 42 – entraînait une détérioration des résultats cliniques des patients.
Dans une étude récente, publiée dans le Journal of Alzheimer’s Disease, nous avons cherché à comprendre ce qui était davantage lié à la progression de la maladie d’Alzheimer : la quantité de plaques dans le cerveau ou la quantité d’amyloïde bêta 42 restante.
Pour répondre à cette question, nous avons étudié les données d’un groupe de personnes porteuses d’une mutation génétique héréditaire rare qui fait courir un risque élevé de développer la maladie d’Alzheimer. Les participants provenaient de l’étude de cohorte « Dominantly Inherited Alzheimer ».
Nous avons découvert que la diminution de l’amyloïde bêta 42 (la version fonctionnelle de la protéine) était plus néfaste que la présence de plaques (les agrégats insolubles d’amyloïde bêta).
Les participants ont été suivis pendant trois ans en moyenne, et nous avons constaté que ceux qui présentaient des taux élevés d’amyloïde bêta 42 dans leur liquide céphalorachidien (LCR) (qui entoure le cerveau et la moelle épinière) étaient protégés. De plus, leur cognition ne se détériorait pas pendant la durée de l’étude. Ces résultats concordent avec ceux de nombreuses autres études qui ont montré l’importance de l’amyloïde bêta 42 pour la mémoire et la cognition.
Ils sont également pertinents parce que nous avons étudié des personnes porteuses de la mutation génétique qui augmente les risques de développer la maladie d’Alzheimer, un groupe qui est considéré comme offrant les preuves les plus solides du caractère néfaste des plaques d’amyloïde bêta. Cependant, même dans ce groupe, ceux qui présentaient des taux plus élevés d’amyloïde bêta 42 dans le liquide céphalorachidien ont conservé des fonctions cognitives normales, quelle que soit la quantité de plaques dans leur cerveau.
Il convient aussi de mentionner que dans certaines formes héréditaires rares de la maladie d’Alzheimer – par exemple, chez les porteurs de la mutation Osaka ou de la mutation arctique –, les personnes peuvent développer une démence avec de faibles niveaux d’amyloïde bêta 42 sans qu’on n’ait pu détecter la présence de plaques. Cela laisse penser que ce ne sont pas les plaques qui sont à l’origine de leur démence, mais plutôt le manque d’amyloïde bêta 42.

Lecanemab – une exception
Comment nos découvertes influenceront-elles la mise au point de médicaments et les essais cliniques pour la maladie d’Alzheimer ? Jusqu’au tout récent essai sur le Lecanemab, un anticorps qui réduit les plaques, tous les essais de médicaments pour la maladie d’Alzheimer s’étaient soldés par un échec.
Certains médicaments ont été conçus pour réduire le taux d’amyloïde bêta 42, en partant du principe qu’en réduisant le taux de la protéine normale, les patients présenteront moins d’accumulations de plaques. Malheureusement, ces médicaments ont souvent empiré l’état des patients.
On a récemment observé que le Lecanemab avait un effet léger, mais significatif sur la réduction du déclin cognitif. Des études antérieures ont démontré que ce médicament augmentait les niveaux d’amyloïde bêta 42 dans le LCR. Cela rejoint notre hypothèse, à savoir que l’augmentation de la protéine amyloïde normale peut être bénéfique.
Nous en saurons plus après la publication des résultats des essais sur le Lecanemab. Pour l’instant, nous ne disposons que d’un communiqué de presse des producteurs du médicament.
Nous pensons que les futurs essais devront se concentrer sur les niveaux d’amyloïde bêta 42, et sur la question de savoir s’il est bénéfique d’augmenter et de rétablir son taux à des valeurs normales plutôt que de chercher à l’éliminer. On pourrait y parvenir en utilisant des protéines semblables à la protéine amyloïde bêta 42 – appelées « protéines analogues » –, mais qui ont moins tendance à former des agrégats que les protéines naturelles.
Cette approche de remplacement actif des protéines pourrait devenir une nouvelle voie prometteuse de traitement contre la maladie d’Alzheimer et d’autres maladies liées à l’agrégation de protéines, comme la maladie de Parkinson et les maladies du motoneurone.![]()
Andrea Sturchio, MD, PhD Student, Clinical Neuroscience, Karolinska Institutet; Kariem Ezzat, Research Scientist, Laboratory Medicine, Karolinska Institutet et Samir EL Andaloussi, Professor, Laboratory Medicine, Karolinska Institutet
Nous remercions les auteurs et The Conversation pour l'autorisation de republication.
